DMFTpack
DMFTpack is the software for DFT+DMFT calculation. Various projection methods [1] and the impurity solvers are available, e.g., iterative perturbation theory (IPT), self-consistent second-order perturbation theory (SC2PT). The interface connecting DFT package, e.g., OpenMX and impurity solvers (CT-QMC) is also provided.
Developer: Jae-Hoon Sim (email: jhsim4279@gmail.com)
Download
DMFTpack.tar.gz
Requirements
Impurity solver
- (Optional) Hybridization expansion quantum Monte Carlo impurity solver:
[To perform the CT-QMC solver, one of the following solvers should be installed: Otherwise, one can use the (SC) 2PT solver, which is implemented in the DMFTpack itself.]
- Implemented in ALPS library http://alps.comp-phys.org
- Implemented by K. Haule at Rutgers University http://www.physics.rutgers.edu/~haule/ (See also to install impurity solver: link)
Eigen (>=3.3)
- C++ template library for linear algebra: matrices, vectors, numerical solvers, and related algorithms. http://eigen.tuxfamily.org
- Since Eigen version 3.3 or later, you can use the BLAS / LAPACK or Intel MKL libraries.
- Using BLAS/LAPACK from Eigen http://eigen.tuxfamily.org/dox/TopicUsingBlasLapack.html
- Using Intel® MKL from Eigen http://eigen.tuxfamily.org/dox/TopicUsingIntelMKL.html
Installation
-
Install pre-requirements. Note that the LAPACK or Intel MKL libraries are optional:
$ sudo apt-get install gcc g++ $ sudo apt install openmpi-common openmpi-bin libopenmpi-dev $ sudo apt install make $ sudo apt-get install liblapack-dev -
Install Eigen library:
$ git clone https://github.com/eigenteam/eigen-git-mirror -
Install DMFTpack code:
$ git clone https://github.com/ElectronicStructureTheory-KAIST/DMFTinOpenMX_dev.git $ cd DMFTinOpenMX_dev/src $ vi makefile (Replace the variables with ${} to the proper values for your system.) $ make all -
(Optional) Install MQEM to perform the analytic continuation:
$ git clone https://github.com/ElectronicStructureTheory-KAIST/MQEM.git
Example
Hubbard model
-
Test calculation can be performed for single-band Hubbard model as following:
$ cd ../example/Hubb_model/primitive_Cell_ipt $ mpirun -np 4 ../../dmft | tee "std.out" $ gnuplot gw_loc_Im.gnuplot $ gnuplot sw_Im.gnuplot -
Now, one may want to do analytic continuation from the Matsubara Green’s function (or the Self-energy) to the real-frequency spectral function.
$ mkdir realFreqSpectrum; cd realFreqSpectrum $ mkdir continuation; cd continuation- Any continuation method can be used to obtain the density of states from the Green’s function, e.g., MQEM (PRB 98, 205102 (2018))
$ cp ../../Sw_SOLVER.full.dat ./ $ julia $(MQEM_dir)/src/mem.jl Sw_SOLVER.full.dat ../../input.solver | tee "std.out"or ΩMaxent (PRE 94, 023303 (2016)).
$ cp ../../Sw_SOLVER.dat ./ $ ~/bin/OmegaMaxEnt | tee "std.out" - For the case of self-energy continuation, one may want to generate “realFreq_Sw.dat_i_j” files, using the Kramers-Kronig relation. Here i and j are orbital indices. Each file contains omega for the first column and real and imaginary part of the self-energy for the second and third column, respectively.
-
(optional) For the system with large spin-orbit coupling, we recommend MQEM method developed by J.-H. Sim [2]. The source code will be available soon via GitHub (update: 2018-10-19).
-
mqem.input.toml file is generated, one can control parameters for MQEM continuation method as following (See https://github.com/KAIST-ELST/MQEM.jl for more details):
-
(key) = (variable)
-
- Any continuation method can be used to obtain the density of states from the Green’s function, e.g., MQEM (PRB 98, 205102 (2018))
-
band structure: To do calculate band structure, SOLVER_TYPE=TB and RESTART>1 in input.pam is required, providing “realFreq_Sw.dat_i_j”.
$ mkdir ../bandstruc; cd ../bandstruc $ cp -r ../continuation/realFreq_Sw.dat_* ../../Inputfiles/* ../../Restart ./ $ vi input.parm $ mpirun -np 4 ../../../../dmft | tee "std.out" -
Now one can plot the band structure as following:
$ python qsband.plot.py
SrVO3
- As same way, one can obtain band structure of the SrVO3 .
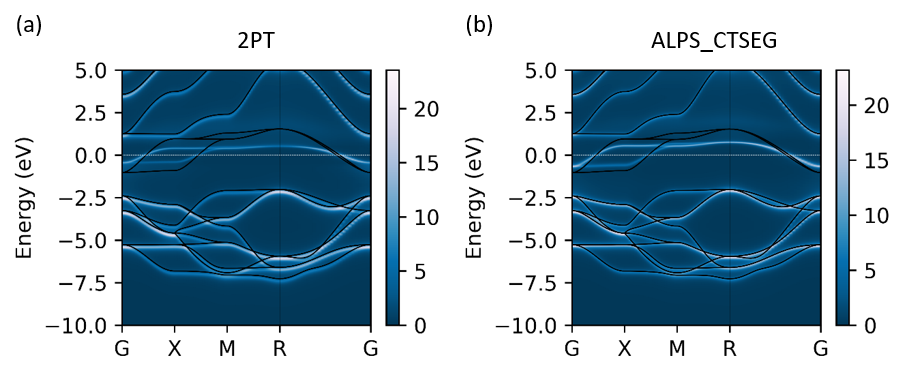
- For DFT+DMFT calculation, non-interacting Hamiltonian is written in “SYSTEM_NAME.scfout” file, which is the output of the OpenMX code. (In this example one can simply uncompress “SVO113.tar.xz” file)
- The figure below shows that the SrVO3 Band structure, calculated by two different impurity solvers, namely 2PT and ALPS_CTSEG.
Document
list of input parameters
After running OpenMX with option “ HS.fileout on”, two additional input files required for DMFT calculation.
-
List of input files
-
“input.parm”: the computational information is written here. See below for more details.
-
“input.solver”: information for impurity solver, e.g., U, N_TAU, …
-
“SYSTEM_NAME.scfout”, which can be obtained from OpenMX code output.
Alternatively, “SYSTEM_NAME.scfout” will be replaced by “Hk.HWR” and “OverlapMatrix.HWR”(optional): non-interacting Hamiltonian is written in the “Hk.HWR” file. When the non-orthogonal basis is used, the overlap matrix should be provided.
-
- input.parm
- Computational information:
- SYSTEM_NAME
- SOLVER_TYPE = {ALPS_CTSEG, RUTGERS_CTSET, RUTGERS_CTHYB, IPT, SC2PT, 2PT, TB}
- SOLVER_DIR = path to impurity solver (default: ~/bin/)
- SOLVER_EXE = executable impurity solver (default: hybridization)
- MAX_DMFT_ITER ={1,2…}, maximum DMFT iteration (default: 20)
- MIXING = (default: 0.8), self-energy mixing weight in the DMFT iteration
- RESTART = {0,1,…}, for initial calculation, >0 to start self-energy obtained from previous calculation. (default: 0)
- Lattice information
- N_ATOMS = {1,2,..}, the number of atoms in the unit cell
- N_CORRELATED_ATOMS = {1,2,…}, the number of atoms containing strong correlated orbitals.
- N_ELECTRONS = {1,2,…} total number of the electrons in the unit cell.
- MAGNETISM = {0,1,[2]} (default: 2)
- K_POINTS = (nk1,nk2,nk3) k-grids to discretize the first Brillouin zone.
- DFT+DMFT option
- H0_FROM_OPENMX = {0,1} = 1 if “SYSTEM_NAME”.scfout file is exist. Otherwise, Hk.HWR file should b exist in the work folder with “H0_FROM_OPENMX”=0.
- num_subshell = the number of nl subshell for each orbital, e.g., num_subshell=5 for s2p2d1 basis
- rot_sym = Quantum number l of the angular momentum for each subshell.
- subshell = dimension of the subshell.
- Rydberg_set = 0 for nominally occupied orbitals, 1 for empty
- Projection method
We provide a method to obtain projective Wannier functions based on the natural atomic orbitals. The basic idea of the method is introduced in Ref. 1.
- MODEL_WINDOW_U = upper bound of the energy window for the correlated subspace. The correlated orbitals are projected into the model window (default: 10).
- MODEL_WINDOW_D = lower bound of the energy window for the correlated subspace (default: -10)
- DC_TYPE = {nominal, [fll]}, double counting method (default: fll)
- N_d = {0,1,..}, nominal charge in the correlated orbitals
- HARTREE_ATOMS = {1,2,…} Atom indices of the correlated atoms
- HARTREE_ORBITALS_RANGE = the range of the correlated orbitals indices
- DOS & Band calculation
- MODE = {band, qsband, dos, qsband}, specifies the band/dos calculation for non-interacting (or quasi-particle) dispersion
- K_GRID_BAND = (default: 40)
- SPECTRAL_ENERGY_GRID = (default: 1000)
- N_K_PATH = the number of paths for the band calculation
- K_PATH = this keword specifies the paths of the band dispersion
- Computational information:
- input.solver
- impurity_information
- N_ORBITALS = the number of orbitals in impurity site
- N_HARTREE_ORBITALS = the number of the correlated orbitals
- U = intra-orbital interaction
- U’ = inter-orbital interaction
- J = Hund’s coupling
- BETA = inverse temperature
- Matsubara_frequency
- N_MATSUBARA = the number of the Matsubara frequencies
- N_TAU = mesh grids in the imaginary time axis
- ALPS_CT_HYB_input
- MEASURE_legendre = should be choose “1”
- MEASURE_freq = should be choose “1”
- TEXT_OUTPUT = should be choose “1”
- MU_VECTOR = should be set as “mu_vector.alps.dat”
- DELTA = should be set as “delta_t.dat”
- impurity_information
Useful output files
-
Gw_loc.full.dat[N]: Local Green’s function Gαβ(iωn) for N-th correlated atom. For each line, five numbers represent the n, α, β, real and imaginary part of the Green’s function, respectively.
-
Gw_imp.full.dat[N]: Same for impurity Green’s function.
-
Sw_SOLVER.full.dat: Self-energy calculated from the impurity solver.
-
Numele.dat: Number matrix nαβ calculated by -Gαβ(τ=β-).
References
- DFT+DMFT with natural atomic orbital projectors Jae-Hoon Sim and Myung Joon Han, (Submitted)
- Maximum Quantum Entropy Method Jae-Hoon Sim and Myung Joon Han, Phys. Rev. B 98, 205102 (2018)